Mission statement: Our aim is to improve computational tools and apply them to biological problems important for improving human health.
Vision: macromolecular interactions are crucial for function. They are exploited for engineering and medical needs. But, computational tools are most useful when these interactions involve limited conformational changes — e.g. in many drug discovery pipelines.
However, most interactions involve large conformational changes. A large fraction of our proteome contains Intrinsically disordered proteins or disordered regions that fold upon binding (e.g. peptide epitopes), protein-nucleic acid interactions often involve large conformational changes, and we are challenged to model them. Our goal is to improve such computational pipelines to increase our capacity to predict molecular interactions.
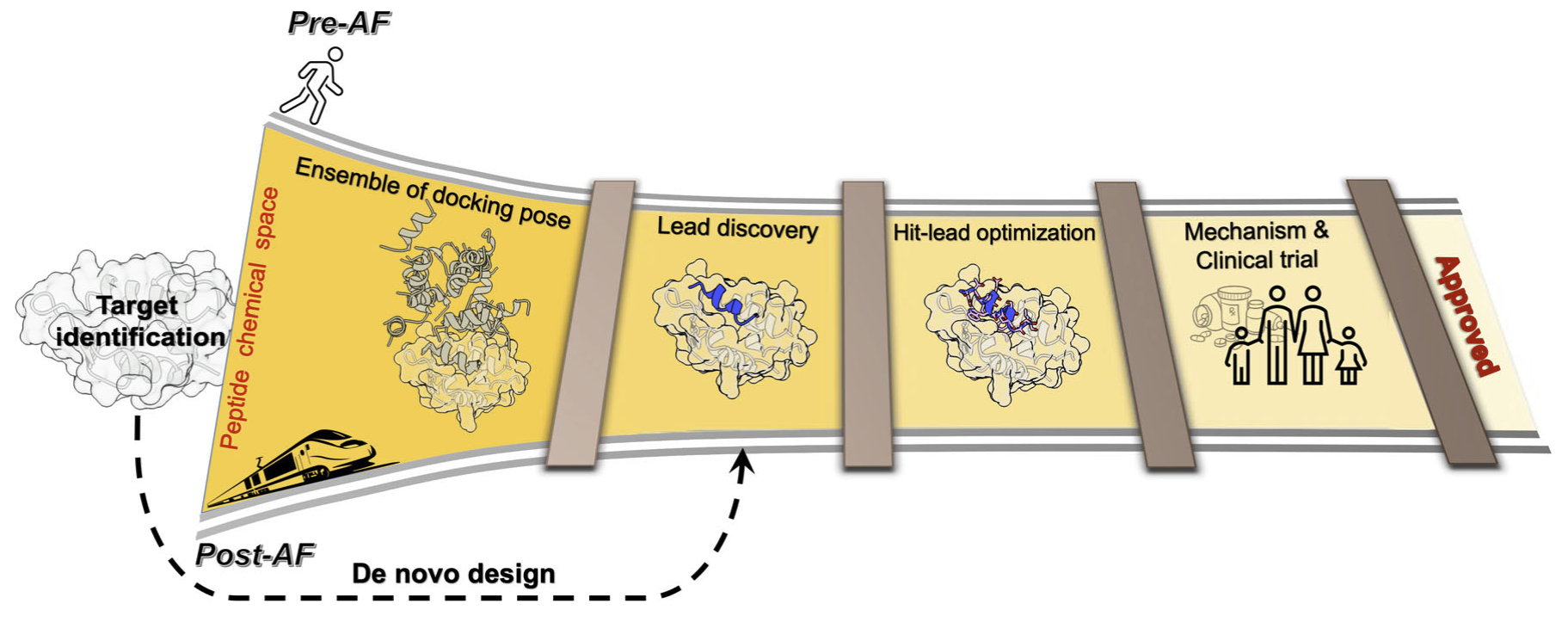
AI is enabling a faster discovery rate
Traditionally we focus on physics-based approaches based on molecular simulations to understand biology — but they are computationally demanding and cannot scale to the number of possible molecules (e.g. sequences) that are biologically relevant. We are leveraging our physics principles to inquire into what AI has learnt about physics, and leveraging this in discovery, for example of novel peptide epitopes. Ultimately, we believe this will accelerate discovery of new molecules for drug discovery.
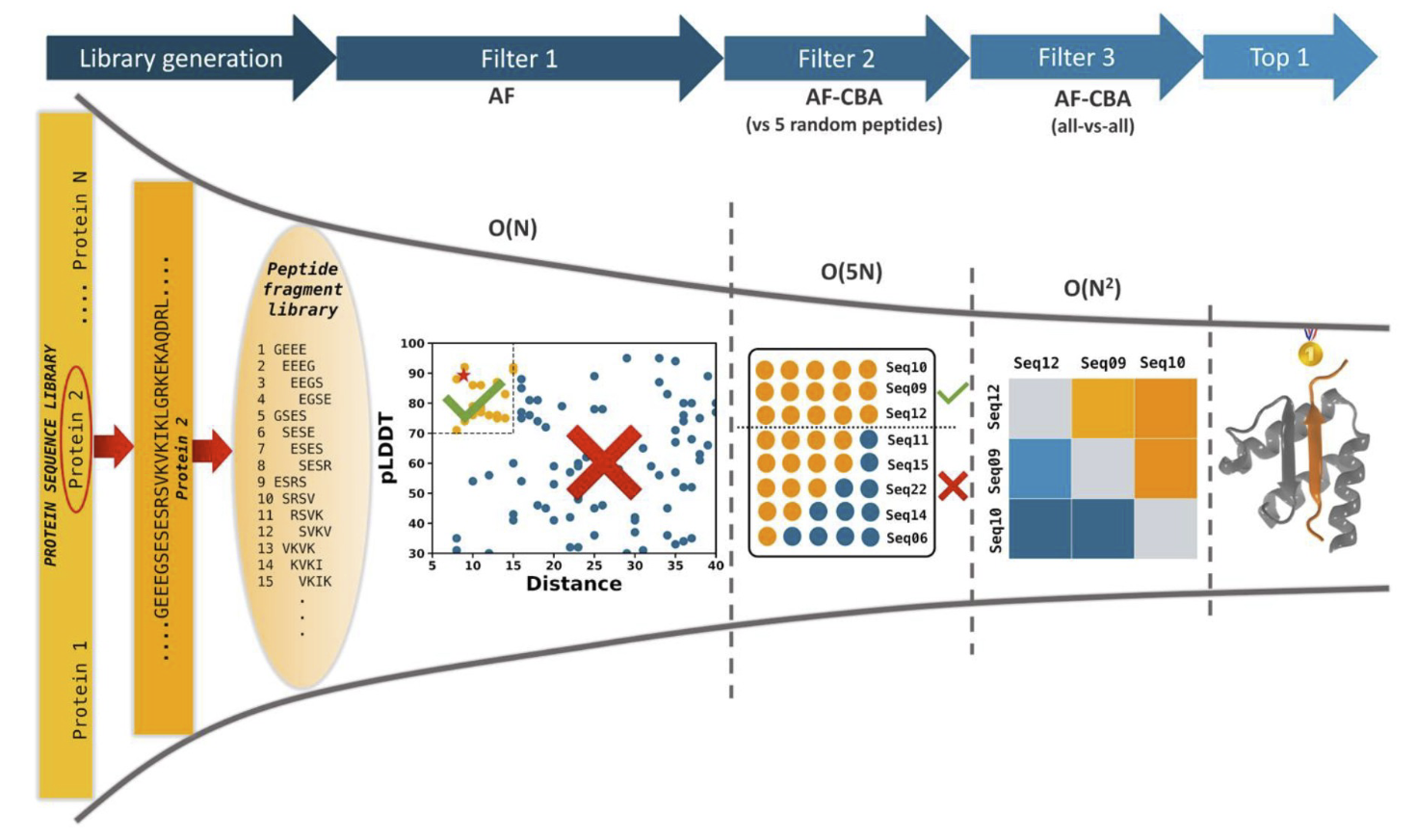
We developed a competitive binding assay using AlphaFold that allows us to identify strong peptide binders by competing pairs of candidate peptides against the same target receptor.
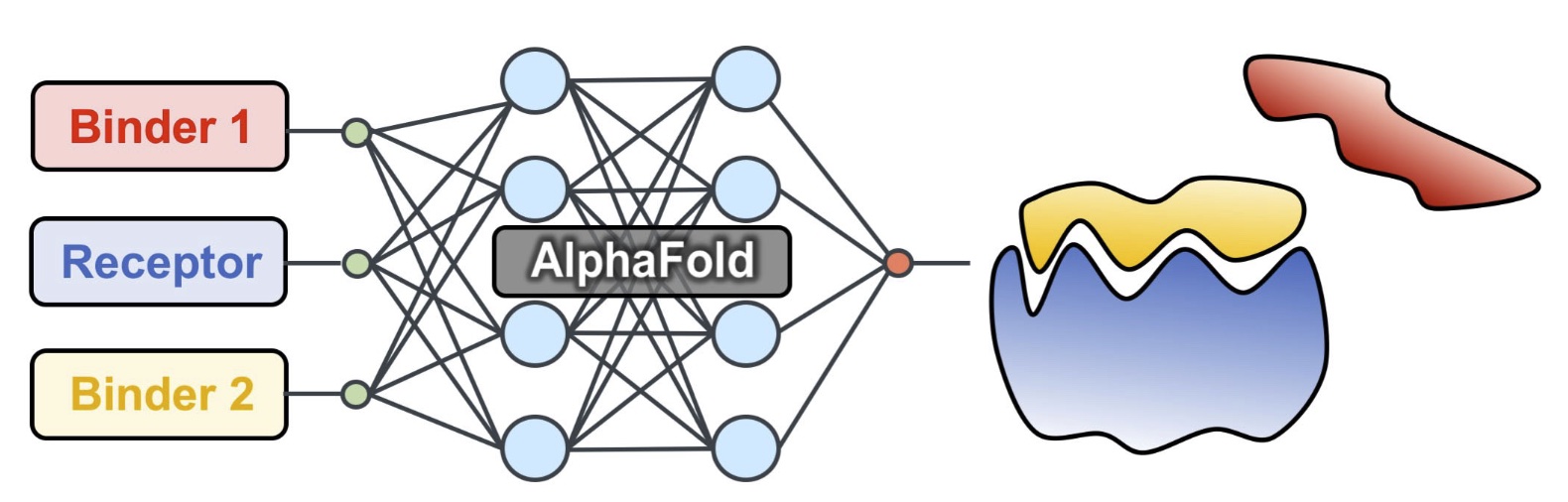
We have used this approach to identify novel peptides that bind the ExtratTerminal domain of BET proteins (involved in immune response) from experimental pull-down experiments — we discovered new motifs, different from known bioinformatics motifs, that have been experimentally verified.
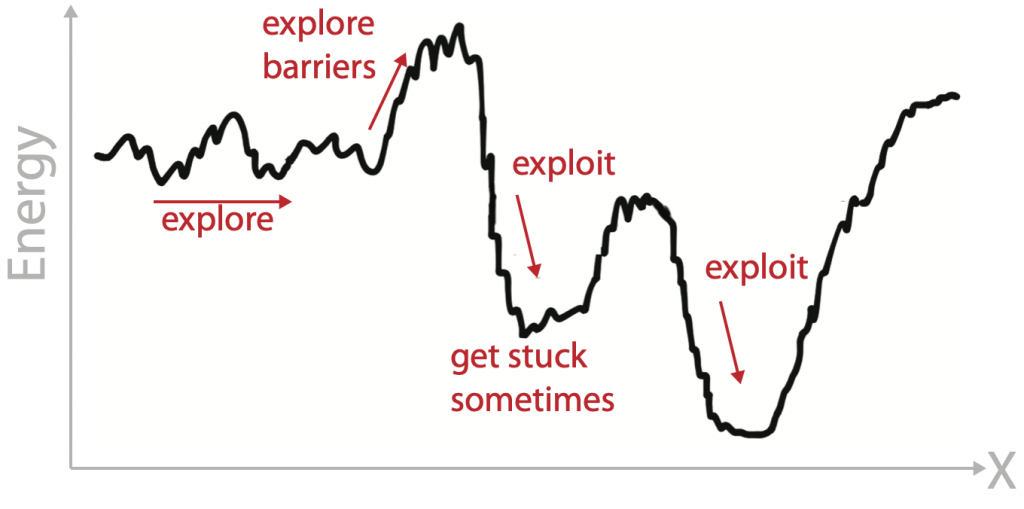
The challenge of sampling energy landscapes
Biomolecules have many degrees of freedom leading to frustrated energy landscape. We used advanced sampling techniques based on molecular dynamics to explore these energy landscapes and exploit regions of interest in it. Depending on the problem at hand using different sampling techniques work best to change the exploration/exploitation trade-off.
Integrative Structural Biology: MELD
There’s an array of biophysical techniques that provide data that is sparse, noisy and ambiguous — not enough by itself to characterize the structures of biomolecules. We have co-developed MELD (Modeling Employing Limited Data) as a plugin to the popular OpenMM and used for problems as diverse as protein folding, binding and structure determination. We plan to continue developing and popularizing this approach.
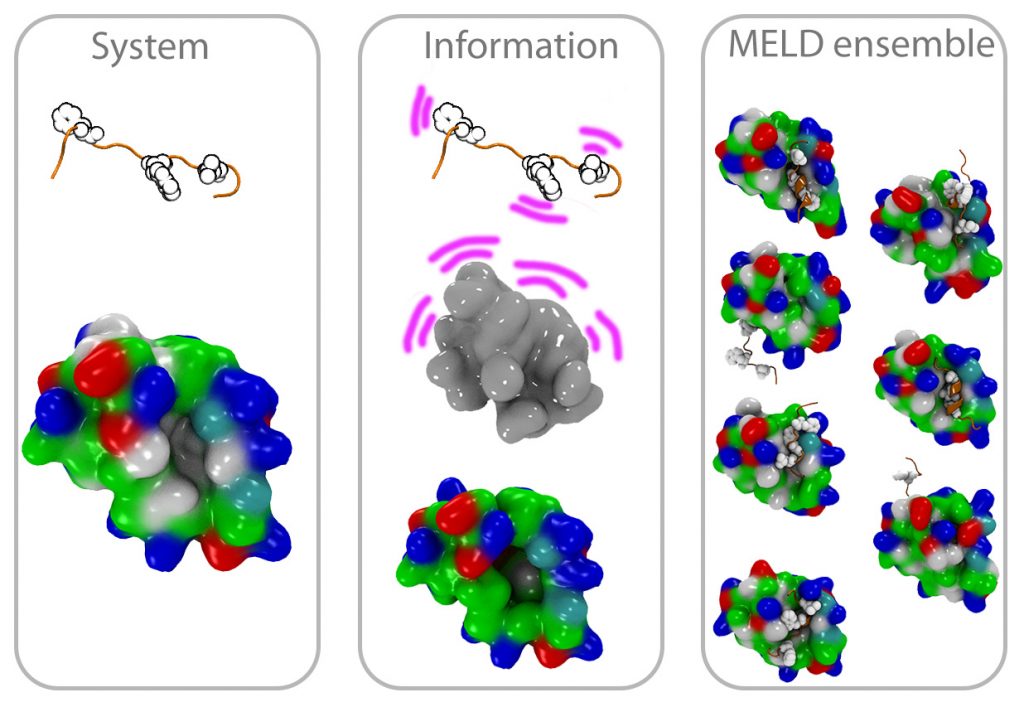
CryoEM structure determination
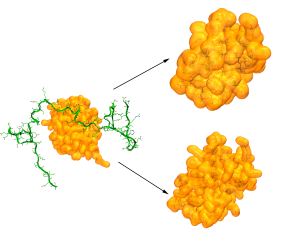
With the resolution revolution has come the need for new tools that can help in determining the structures of biomolecules and their assemblies. We have been collaborating on a pipeline combining MDFF and MELD that produces high accuracy structures from different resolution maps. By using physical models to guide the sampling we overcome issues such as dependency on good secondary structures. At the same time MDFF accelerates the sampling towards structures that fall within the electron density map.
NMR structure determination
NMR provides an array of different tools to understand biomolecular structure and dynamics. NMR data can provide information about conformations for bonded terms or even provide proximity information for atoms far in the sequence (e.g. NOESY). With thousands of data points, the commmunity can solve the structures of proteins with thousands of atoms. We been working within the context of CASP to develop better methodologies to solve structures when the information is more sparse. Combined with selective isotope labeling this can help scale NMR approaches to larger proteins that can currently be solved.
At the other end, we are interested in the binding of peptides to proteins. Isotopically labeling peptides is expensive, limiting the use of NMR when multiple sequences are of interest. However, using Chemical Shift Perturbation on the protein spectra can help identify possible regions of interaction between the peptide and protein, which combined with our MELD approach can yield atomistic detail.
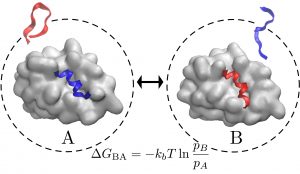
Binding affinity and kinetics
Current drugs target a subset of the proteins directly involved in disease. Many of these proteins remain undruggable because drugs need of a foot-hold or cavity in which to bind. Peptides on the other hand are much more suited to interact with those ‘undruggable’ proteins — but current tools are not well suited for that mission. On top of the traditional problem, peptides exhibit high flexibility. We have shown an efficient approach to identify binding poses and even relative binding affinities between similar peptides sequences. We want to further use Markov State Models to characterize kinetic properties such as kon and koff. koff has been shown to be particularly important to control the time the peptide interacts with its partner protein and it is difficult to calculate experimentally.
Gene regulation rules
Projects like ENCODE have brought a wealth of sequence information. We are combining this sequence information with structural insights from binding simulations to understand how transcription factors determine specificity. During the binding process some DNA can deform significantly from their canonical B-DNA conformation.
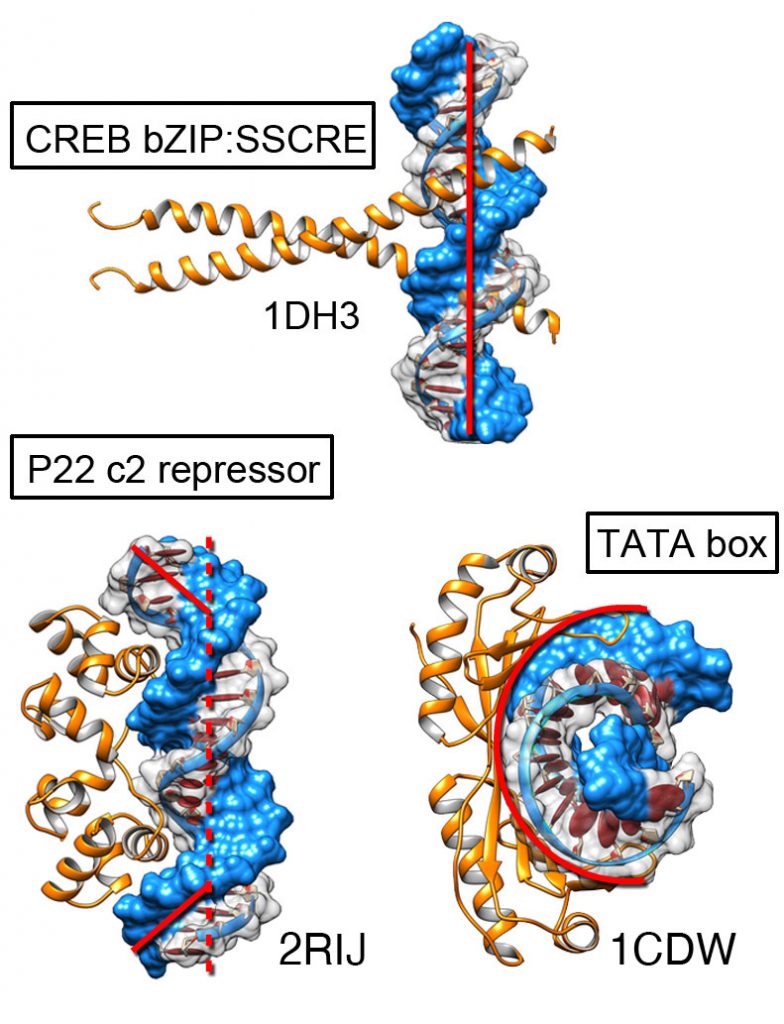
A key challenge with proteins that bind nucleic acids is that a single transcription factor might bind to different DNA sequences with good affinity. Hence, it is the relative binding affinity that is important. Traditionally we distinguish between proteins that interact through sequence specific contacts vs those that bind to general features of the DNA (e.g. phosphate backbone). Some DNA sequences can deform to bound conformations at lower energetic cost, favoring interaction (shape readout). We use our tools to identify the balance between direct interactions and shape complementarity.
We are part of the Ascona B-DNA Consortium (ABC for short) which consists of several groups interested in molecular simulations of DNA who propose state-of-the-art simulation protocols for the study of nucleics and provide datasets of all possible n-mers of size n. The previous ABC set simulations of all possible tetramers and in 2021 we are starting a new round of simulations to scale beyond. We have used our previous datasets in collaboration with Remo Rohs to look predict TF binding preferences.